Advertisement
FDA Proposes Tougher Rules For Surgical Mesh
The U.S. Food and Drug Administration is proposing tougher regulations for vaginally implanted surgical mesh to treat women with prolapse, The Wall Street Journal reports.
In a document posted to FDA's website Thursday the agency said, the "rate and severity of mesh-specific adverse events following vaginal POP [pelvic organ prolapse] repair with mesh calls into question the safety of these devices." Many side effects were caused by the mesh protruding out of the vaginal tissue, which in turn caused pain, infection and bleeding.
The agency said is proposing new mesh products undergo a premarket review process, which requires companies to conduct studies looking at a product's safety and effectiveness prior to approval. The mesh products are currently reviewed under the less stringent review known as the 510(k) process. The agency is also proposing that studies involving current products be conducted but said it would consider a grace period for companies to submit a premarket approval applications.
An FDA advisory panel is being called on to discuss the mesh products next week. The meeting [Sept.8-9] is expected to mostly focus on mesh when used to treat organ prolapse, but the panel will be asked to discuss the use in treating a condition known as stress urinary incontinence. The consumer group Public Citizen petitioned the FDA last week to recall existing mesh products and require new products to undergo a premarket review process.
In a piece yesterday, I wrote about a group of pelvic surgeons who are challenging some of the FDA's safety warnings.
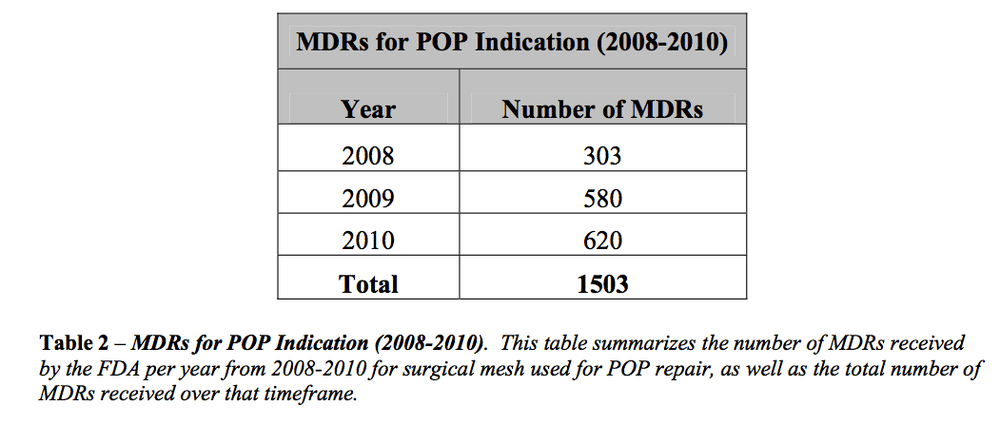
This program aired on September 1, 2011. The audio for this program is not available.
