Support WBUR
In Step Toward Genetic Fix, Scientists Pinpoint 'Achilles Heel' Of Sickle Cell Disease
It’s not like any other pain, says Cloret Carl of Quincy. It’s inescapable, and far beyond a toothache or childbirth or a joint replacement: “It encompasses every single part of your being and your body, because it’s in your blood,” she said.
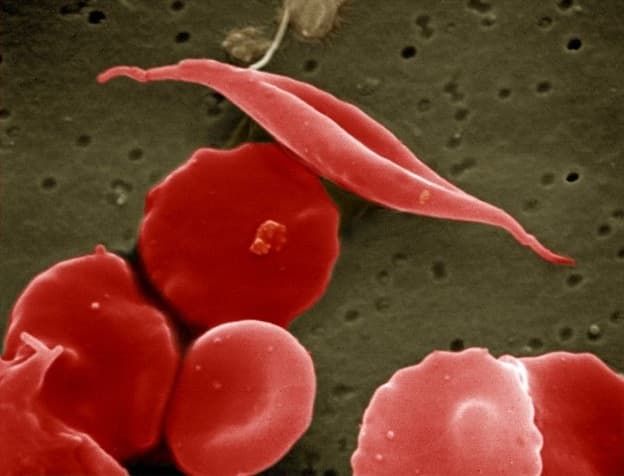
She’s describing the periodic agony of sickle cell disease, a genetic disorder that mutates red blood cells from their usual disc shapes into sickles, leading to dangerous — and excruciating — blockages. Organ damage and early death can result.
Carl, who supports the Greater Boston Sickle Cell Disease Association and works with doctors and researchers, has lost friends young and old to sickle cell disease, and spent many days in intensive care units with life-threatening complications herself. Sickle cell disease affects about 100,000 Americans, mainly of African descent, and though there has been progress on treating it in recent years, “most of the people who have this illness have a very painful existence,” she said.

So she’s heartened by the latest findings on a tantalizing prospect for a possible eventual cure: attempts to turn back the genetic clock on sickle-cell disease.
“This gives hope to people that at some point, we’re going to conquer this illness,” she said.
The concept: Hemoglobin, which carries oxygen, is mutated in the disease. What if scientists could reactivate long-dormant genes to replace the mutated hemoglobin? What if they could spur patients to produce a healthy form of hemoglobin that their bodies stop making soon after birth?
That prospect came one step closer this month with the publication of findings in the journal Nature that pinpoint a narrow DNA target for such a fix.

“We located a very, very small region, basically an Achilles heel, that we think would be very amenable to editing and to therapy,” said Dr Stuart Orkin of the Dana-Farber/Boston Children's Cancer and Blood Disorders Center, a senior author on the paper.
Biotech companies hope to begin trying gene therapy on this new target in patients as soon as next year, he said.
Sickle cell disease is caused by a single mutation, so of course the most logical way to cure it would be to simply repair that mutation, said Dr. Daniel Bauer, also of the Cancer and Blood Disorders Center and a senior author on the Nature paper. But it’s much harder to fix a genetic sequence than it is to just break one, he said.
So precision breakage became the goal. But was it possible?
“What we didn’t know before this study was, would it be possible to break the target we wanted to break with a single cut” of DNA, he said. “What this study revealed is yes, we can do it, and it’s actually a very potent strategy.”

Their target was, in effect, a genetic “off” switch. In the womb and early infancy, we produce a fetal form of hemoglobin, but then, in early life, we stop, switching almost entirely to an adult form. That’s fine, unless you’re a sickle cell patient whose adult form of hemoglobin is perilously misshapen.
It has long been known that the fetal form can successfully replace the adult form, Orkin said. In adults with hemoglobin disorders, “this should, in fact, essentially cure them,” he said.
But how to do it? Several years ago, Orkin and other researchers had found a “switch factor” that shuts off the fetal form. So how to flip this “off” switch back to “on”?
Orkin and Bauer’s research team surveyed the DNA neighborhood, testing more than a thousand spots. “We needed to locate a place in the switch gene that really would be convenient to the new kind of gene-editing tools,” Orkin said.
Those tools include the sizzling biotechnology known by the acronym CRISPR, a method that makes it so much easier to edit DNA that it has been compared to switching from a typewriter to a word processor.
"I think there’s a lot more optimism now than there was in the past in terms of better, even curative options for these disorders."
Dr. Daniel Bauer
Their Nature paper pinpoints the best spot. And gene editing tools would also help convert their findings to treatments, they say.
The plan, Bauer said, would be to extract blood stem cells from a patient, manipulate the cells’ DNA to turn the fetal hemoglobin back on, and then put those altered cells back in the patient. “The patient’s blood system would then be maintained by cells that were corrected,” he said.
“This is really a type of therapy that we could only imagine in recent years, when the genome-editing technology has become practical,” he noted.
Work continues on how to target the edits as effectively as possible, he said, “only editing the spot we want to edit, and not causing any unwanted changes.”
But Orkin said that with the key DNA region identified, a practical way forward is now clear.
With many discoveries, he said, several more advances are needed before the science can possibly be of use to patients. “Here, there’s no more real unknown science,” he said. “It’s really just a question of translating it.”
Boston Medical Center hematologist Dr. Martin H. Steinberg, who has researched sickle cell disease and treated patients for decades, agreed that the paper details a potentially very useful target.
A few hundred sickle cell disease patients have been cured by bone marrow transplants, he said, but many are not eligible or lack compatible donors. If a patient’s own blood cells could be re-engineered, that would eliminate the need for a donor and concerns about rejection.
“But it’s not going to happen tomorrow,” he cautioned, and the process would be labor-intensive and require an advanced hospital. “If you’re asking for a time, I would say you’re looking at years,” he said. “Five, 10 years, I don’t know.”
Though he praised the paper, which he was not involved with, and its authors, “it’s a long ways to go from these observations to something that’s therapeutically available.” There are many “technical impediments” to this and other gene therapy work, he said.
Bauer agreed that “we’re not there yet.” He said treatments based on the gene-switch work “will be introduced in very careful, small clinical trials with close observation, so it’s not going to be ready for prime-time for every patient immediately.”
“But,” he said, “I think there’s a lot more optimism now than there was in the past in terms of better, even curative options for these disorders.”
Sickle-cell disease research — or rather, the paucity thereof — has long raised questions of racial discrimination.
Consider, for example, this recent Mother Jones story: “One Disease Hits Mostly People of Color. One Mostly Whites. Which One Gets Billions in Funding?” That translated into a Daily Kos headline: “How Race Plays An Ugly Role in the Drastic Underfunding of Sickle Cell Research and Advocacy.”
And Forbes ran this headline this summer: "Sickle Cell Disease Highlights Racial Disparities In Healthcare."
The story notes:
“The funding disparities for research on sickle cell compared to other pediatric diseases are huge. Cystic fibrosis, a disease that affects primarily Caucasians, occurs in only a third of the numbers affected by SCD, but received 3.5 times more NIH funding. Private funding from foundations was about 400 times higher for cystic fibrosis! Unsurprisingly, Johns Hopkins researchers John Strouse and Carlton Haywood note that no drugs were approved between 2010 and July 2013 for the treatment of SCD compared with five for cystic fibrosis.”
Cloret Carl of Quincy agrees that compared to funding for research on other diseases, sickle cell disease is dramatically underfunded. She said people in the sickle cell community also report stigma — but mainly in connection with seeking treatment for the disease’s pain crises.
“The big stigma is that people with the illness are drug seekers,” she said. In emergency rooms, she said, “The first misjudgment is, ‘Oh, this person is seeking narcotics,’ because it takes high dosing of pain medication administration for you to get that breakthrough,” for the pain to start to resolve.
And the argument that sickle cell disease research is underfunded because it mostly affects people of color?
Sickle cell disease affects Asians, Africans, people of Italian and South American descent, Carl said; perhaps it’s more about “people being poor.”
