Support WBUR
How A Woman In Her 70s Might Be The Key To A New Alzheimer's Treatment
Normally, everyone with a certain mutation on a gene called PSEN1 begins experiencing the first symptoms of Alzheimer’s disease around their mid 40s.
“Close to 100% will develop dementia," said Yakeel Quiroz, a neuropsychologist at Harvard Medical School. "The diagnosis usually comes around age 49.”
Everyone except one patient, that is. Quiroz and her colleagues studied this woman, who didn’t develop dementia until her 70s, despite carrying the mutation. Researchers first realized she carried the mutation, known as E280A, when she joined an Alzheimer’s study in 2010. Quiroz said she couldn’t believe it at first.
“We ran the genetics like three or four times just to make sure she really had it,” she said. “You wouldn’t suspect it at all. For instance, she was really good at remembering conversations, and what she did the night before. It was really impressive!”
Typically, people with dementia don’t have that level of lucidity, Quiroz said.
Researchers also noticed that this patient has another mutation – something called the Christchurch mutation – in a different gene called APOE.
It appeared that this second mutation “acts like a shield” against early-onset Alzheimers, said Joseph Arboleda-Velasquez, Quiroz's colleague and a biologist at Harvard Medical School.
He suspects this mutation somehow allowed the patient to remain healthy for decades, and he and Quiroz say that understanding why might lead to treatments for Alzheimer’s patients in the future.
WBUR spoke with Arboleda-Velasquez and Quiroz about the discovery, which they reported Monday in the journal Nature Medicine. The interview has been edited for length and clarity.
What were the first things you noticed about this patient?
Quiroz: She’s in her 70s and from Colombia. A research group there began recruiting for a clinical trial around 2010, and they invited people with a family history of early onset Alzheimer’s disease. Her family has that. That’s how they found out she was positive for the E280A mutation we were studying for early onset Alzheimer’s disease.
When we found out about her, we arranged for her and her family to come to Boston. First it was pretty much to characterize the pathology of the disease – so was there anything about her brain that was different?
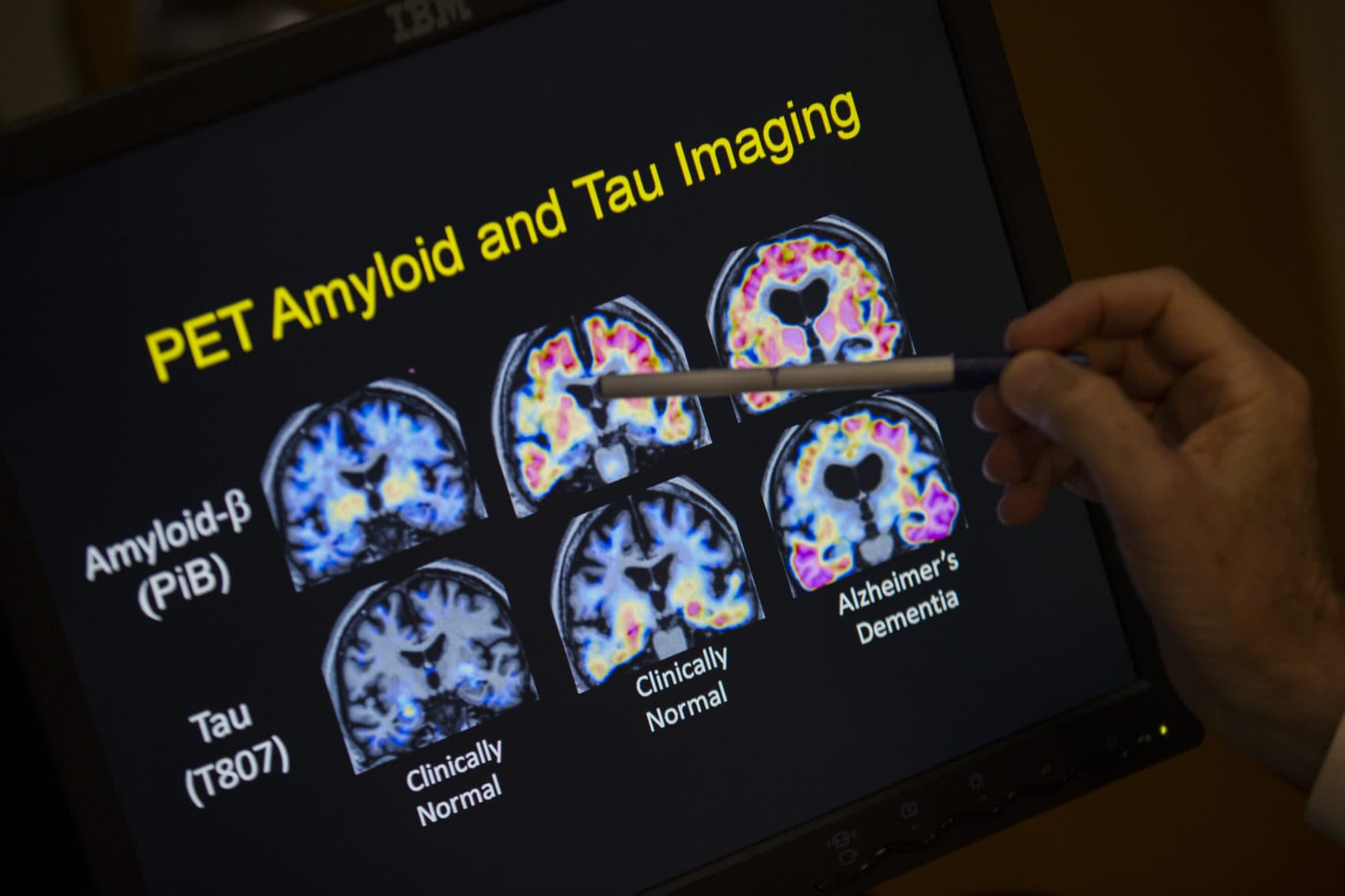
One of the first things we noticed from the PET scan was she had a lot of amyloid protein.
Arboleda-Velasquez: Overproduction of amyloid can trigger the disease of Alzheimer’s at an earlier age. That’s what happens with this E280A mutation. Too much amyloid, and it can form clumps that become toxic. It has the potential to kill neurons, and it causes other problems in another protein called tau that also starts to aggregate and become toxic. So, it’s a cascade of bad things that starts with amyloid.
Quiroz: That was one of the most surprising moments. Even in the presence of a lot of amyloid, her brain was still resistant. Usually, a lot of potential Alzheimer’s therapies try to reduce amyloid. But here it was like, look! She has a lot of it but didn’t develop the tau pathology and is unimpaired.
How come this overproduction of amyloid wasn’t affecting her the same way it did for other people with the same mutation?
Arboleda-Velasquez: So she has two copies of this Christchurch mutation in the APOE gene. This gene makes a protein called Apolipoprotein E. What we found is that when someone has two copies of this gene variant, that person will not develop Alzheimer’s in her 40s if she was predisposed to develop the disease. APOE has a lot of functions in immunity and fat metabolism and people have known for decades that this gene is very important for Alzheimer’s disease risk. The Christchurch mutation stops the APOE protein from binding to a sugar called heparin.
That seems to be the glue that was connecting a lot of pathological effects of Alzheimer’s disease. Once you have a version of APOE that is unable to bind heparin, the whole cascade just falls apart. It could be that the way amyloid is also forming in the presence of this gene is not that toxic, or the mutation is protective because the disease just can’t progress. What the finding is really telling us is that this is potentially very important, and we need to focus on better understanding how it works. It might have a significant impact on treatment for patients in the future.
How does this help find a treatment for Alzheimer’s?
Quiroz: In the paper, we mention an antibody that could mimic the function of the Christchurch mutation between APOE and the heparin sugar. If the antibody can interfere with the binding between APOE and the sugar, then it might not matter how much amyloid your brain is producing. Your brain can be resistant and give you some resistance to dementia. It’s a way that we can develop a drug. We’re doing lab work right now trying to see if this can be done.
The next step is to test it into an animal model like mice, and see if it can make it through clinical trials at some point. What I do want to emphasize is that everything we are learning here we found through one individual. That’s how much info one case can provide.
